Day 2 :
Keynote Forum
Ahmed Radwan
Pioneera Health Care Group, UK
Keynote: Biosimilars: Expanding the scope of biologic therapy through wider patient access
Time : 10:00-10:40

Biography:
Ahmed Radwan has received his MSc and Doctoral degrees from Middlesex Business School in London. He also holds a first degree in Pharmacy and Pharmaceutical Sciences from Cairo University. He is an Adjunct Professor of Management at Edinburgh Business School, Herriot Watt University in the UK. He is also the Chairman and CEO of Pioneera HealthCare Group. He has over 25 years of experience within the pharmaceutical sector, including ethical pharmaceuticals, OTC, generics and strategic management consultation in the pharmaceutical field. He was the Marketing Director of Novartis, Regional Marketing Director of Otsuka and Regional Marketing Director of GSK.
Abstract:
Background: Medicinal compounds are often organic compounds, which are divided into classes of small organic molecules and biologics. Chemical drugs are made of pure chemical substances and their structures can be identified. Biologics are made from natural sources and are used to treat medical conditions. Most biologics, however, are complex mixtures that are more difficult to identify. They have larger molecules than small molecule drugs. "Biosimilar" is a term that is used for copy versions of biologics. There is more variation when trying to replicate a biologic drug than for drugs made by chemical synthesis, so, the term "biosimilar" is used instead of "generic". Biosimilar must have no clinically meaningful differences between it and the reference product it was compared to in terms of the safety and potency. Issue: The clinical applications of biosimilars remain under-recognized by several physicians across the globe.
Objective: The aim of the research is to provide a comprehensive review of biosimilars and its potential therapeutic applications in clinical practice.
Methods: Current literature were analyzed, summarized and interpreted to present biosimilars clinical applications.
Results: Some biologics will lose patent protection during the coming few years and could be replaced with lower cost biosimilars. Regulatory guidelines for development and approval are becoming more rigorous. As a prerequisite, the approval process requires solid demonstration of comparability in quality, efficacy, and safety between the biosimilar and the reference
product.
Conclusions: A great potential still exist for biosimilars to expand biologic therapy through better patient access across many therapy areas. A much wider access compared with biologics due to better affordability could be a clear advantage for a biosimilars in the near future.
Keynote Forum
Alexander Heifetz
Evotec (UK) Ltd, Uk
Keynote: Accurate assessment of protein-ligand interaction energy in seconds with quantum mechanics
Time : 10:40-11:20

Biography:
Dr. Alexander Heifetz is Principal Scientist at Evotec (UK) Ltd, a drug discovery services company and visiting scientist at University College London at the group of Prof Andrea Townsend-Nicholson. Alex has more than 15 years of experience in drug discovery industry and was involved in discovery of 4 clinical drug candidates for treatment of anxiety, major depressive disorder, pulmonary hypertension, and Alzheimer’s disease. He has more than 35 patents and 40 publications in the area of medicinal chemistry. In 2011 he received the Royal Society Industry Award for development of methods for GPCR modeling. In 2001 he obtained his PhD from the Weizmann Institute of Science.
Abstract:
The understanding of binding interactions between any protein and a small molecule is a cornerstone of any efficient structure-based drug design (SBDD) process. X-ray crystallography and homology modeling are the main source of structural information required for rational SBDD. However, even with the crystal structure in hand, visual inspection and force field-based molecular mechanics calculations often used for the rationalization of ligand-protein potency cannot always explain the full complexity of the molecular interactions. Quantum mechanical (QM) approach was always considered as promising direction to achieve this goal, however; traditional QM is not feasible for large biological systems, due to their high computational cost. FMO method offers a considerable computational speed-up over traditional QM methods. One of the key features of the FMO approach is that it can provide a list of the interactions formed between the ligand and the receptor and a chemically intuitive breakdown of these interactions. Such information is essential for medicinal chemists to be able to rationally approach modification of lead compounds in order to increase favorable interactions. Recently, we have demonstrated that FMO can be even faster (sec instead of hours) without compromising the accuracy of the calculations by combining it with density-functional tight-binding (DFTB) method. We will demonstrate the prospective application of FMO method in drug-discovery programs and in study of ligand-receptor residence time.
Keynote Forum
Olga I Lavrik
Institute of Chemical Biology and Fundamental Medicine, Russia
Keynote: Synthesis and biological evaluation of novel classes of tyrosyl-DNA phosphodiesterase 1 inhibitors as anticancer drugs
Time : 11:35-12:15

Biography:
Olga I Lavrik has completed her graduation from Novosibirsk State University and post-doctoral studies and PhD from Institute of Bioorganic Chemistry in Moscow. She is the director of Laboratory of Bioorganic Chemistry of enzymes in the Institute of Chemical Biology and Fundamental Medicine of Russian Academy of Sciences. She is the professor of Novosibirsk State University. She has worked as a Visiting Professor at NIEHS (NC), Institute Jacques Monod, France and University of Évry Val d'Essonne, France. She has published more than 300 papers in reputed journals and has been serving as an Editorial Board of Journal of Molecular Biology and Biochemistry, Journal of Molecular Biology and as a Reviewer of Nucleic Acids Research, Journal of Medicinal Chemistry, Journal of Biological Chemistry, Oncology Letters and other journals. She is a Member of Study Section of Russian Fund for Basic Research and Member of Federation of Biochemical Society. She is a Chancellor of European Environmental Mutagenesis and genomics Society. She is a State prize winner and correspondent member of Russian Academy of Sciences.
Abstract:
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a promising target for antitumor therapy based on Top1 poison-mediated DNA damage. TDP1 plays an important role in removal stalled Top1-DNA covalent complexes, generated by DNA topoisomerase I (Top1) inhibitors, such as camptothecin and some other anticancer drugs. A mutation or genetic inactivation of Tdp1 can hypersensitise cells to camptothecin, whereas over-expression of the active Tdp1 protein has been shown to result in a significant reduction of camptothecin-induced DNA damages. Hence, inhibiting the activity of TDP1 can enhance the therapeutic effect of Top1 modulators for anticancer treatment. The row of novel types of compounds belonging to the different chemical classes were synthesized and tested as TDP1 inhibitors using an original oligonucleotide-based fluorescence assay. Some of them surpass world counterparts in efficiency in dozens of times. The study of cytotoxicity of these compounds revealed that all compounds possess moderate to low cytotoxicity. The absence of cytotoxicity is an advantage when used in combination with clinical Top1 inhibitors. Several inhibitors enhance cytotoxicity of camptothecin against tumor cell lines, sensitizing cells to its effects. We have chosen the least toxic inhibitors of Tdp1, possessing sensitizing effect, to test in vivo.We used mice with Lewis carcinoma to investigate the ability of Tdp1 inhibitors to sensitize tumor to the effect of topotecan (clinical camptothecin derivative). When using a combination of topotecan and Tdp1 inhibitor the primary tumor weight decreased by 30% and the number of lung metastases by 70% compared to a monotherapy with topotecan. Therefore, several new classes of very effective inhibitors of TDP1 were elaborated which are very prominent to improve cancer therapy based on TOP1 poison–mediated DNA damage.
- Synthetic Organic Chemistry|Anticancer Agents in Medicinal Chemistry|Bioorganic and Medicinal Chemistry

Chair
Maia Merlani
Tbilisi State Medical University, Georgia

Co-Chair
John Yuen
The Hong Kong Polytechnic University , Hong Kong
Session Introduction
Maia Merlani
Tbilisi State Medical University, Georgia
Title: 5-Steroidal hydrazones: Synthesis and biological activity
Time : 12:15-12:40

Biography:
Maia Merlani has completed her PhD from Tbilisi State University. She is a senior research scientist at Tbilisi State Medical University. Her field of interest is a chemistry and synthesis of biologically active compounds. She is the author of more than 50 papers in reputed journals and presentations at 60 international scientific conferences. She was granted Georgian Presidential scholarship for young scientists (1997), NATO scholarship (2002, 2003) and Matsumae International foundation scholarship (2013). She is a member of organizing committee of several international conferences in the field of organic and pharmaceutical Chemistry.
Abstract:
Several new 5α-steroidal hydrazones have been synthesized and examined for their biological activities. The starting compound for the synthesis was tigogenin, isolated from the plant Jucca gloriosa (family Liliaceae) introduced in Georgia. The condensation of 3β-hydroxy-5α-pregn-16-en-20-one with isonicotinoylhydrazide or p-nitrophenylhydrazine at room temperature in ethanol in the presence of a catalytic amount of glacial acetic acid gives corresponding hydrazone, whereas treatment of the same ketones with phenylhydrazine, p-bromophenyl-, p-chlorophenyl- or p-methylphenylydrazine in the same conditions produces just product of their intramolecular cyclyzation – pyrazolines. Obtained steroidal isonicotinoylhydrazones readily cyclizes on heating to 5α-androstano- [17,16-d]-pyrazolines, whereas in the same conditions cyclization of p-nitrophenylhydrazones did not proceed. It can be explained by the effect of electron withdrawing substituent at the amine nitrogen atom of the hydrazone fragment of p-nitrophenylydrazones, which prevents process of their cyclization. Reaction of epiandrosterone and 5α-androstan-3,17-dione with isonicotinoylhydrazide, m-nitrobenzhydrazide or m-bromobenzhydrazide yields corresponding hydrazones. All synthesized hydrazones and pyrazolines have been examined for their biological activities. Isonicotinoylhydrazone of epiandrosterone revealed high antituberculousis activity against M.tuberculosis H37Rv and can be considered as promising antituberculosis agent. Bis-hydrazones of 5α-androstan-3,17-dione showed high anticancer and anti-HIV activity.
Abdulmajeed S H AlSamarrai
Department of Chemistry, University of Samarra, Iraq
Title: Synthesis and characterization of 2-((4R,4aR,5aS,6S)-1,3-dioxo3m3a,4,4a,5,5a6,6a-octahydro-4,6- ethenocyclopropa[f]isoindol-2(1H)-yl)-N-(substituted phenyl) acetamides anticipated to have HIV- 1 activity
Time : 12:40-13:05

Biography:
Abdulmajeed S H AlSamarrai was awarded a BSc in chemistry by the Mosul University of Iraq and MSc in analytical chemistry , by Baghdad University of Iraq and was awarded a PhD in organic chemistry by university of Birmingham/ UK in 1984. He is an assistant professor, department of chemistry, Tikrit university , Iraq (1991-2001). Then since 2001, he is a faculty member in the department of chemistry at the University of Samarra, Iraq and he is working as a professor of organic chemistry. Dr. ALSamarrai research interests are on organic synthesis of heterocyclic compounds and has published about 30 papers in reputed journals.
Abstract:
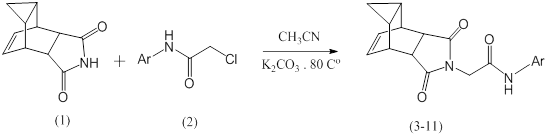
This work includes the synthesis of some new 2-((4R,4aR,5aS,6S)1,3-dioxo3,3a,4,4a,5,5a6,6a-octahydro-4,6-ethenocyclopropa[f]isoindol-2(1H)yl)-N-(substituted phenyl) acetamides (3-11) by reaction of (4R,4aR,5aS,6S)-1,3-dioxo3,3a,4,4a,5,5a,6,6aoctahydro4,6ethenocyclopropa[f]isoindole1,3(2H,3aH)-dione (1) with 2-chloro-N-(substituted phenyl) acetamides (2) in the presences of potassium carbonate in refluxing acetonitrile as a solvent. These compounds have been identified by spectroscopic methods including, IR, 1H-NMR, 13C-NMR and elemental analysis (CHN).
Hasnah Osman
Universiti Sains Malaysia, Malaysia
Title: Identification of thiadiazinyl-coumarin derivatives as dengue virus-2 NS2B/NS3 protease inhibitors
Time : 14:20-14-45

Biography:
Hasnah Osman has graduated with bachelor of science in chemistry from Universiti Sains Malaysia in 1985. She obtained her Master’s degree in 1998 and was appointed as a Lecturer in the year 2000. She completed her PhD in the field of synthetic organic chemistry from University of Otago, New Zealand in 2005. She was promoted to Senior Lecturer in the year 2007, associate professor in 2009 and then became a professor in the year 2014. She is a Member of the Malaysian Institute of Chemistry (since 1998), the American Chemical Society (since 2008) and the Royal Society of Chemistry (since 2009). Currently, she is an ExCo member of the Malaysia Natural Product Society and a Secretary of the Malaysian Ionic Liquid Society. In term of research, she has published more than 150 papers in reputed journals and has been serving as an Editorial Board Member of repute.
Abstract:
Dengue is a dreadful arboviral disease which has become a major public health concern especially in tropical and sub-tropical regions. Despite several reports of peptide based dengue inhibitors, recently the development of small molecules against dengue has gained tremendous momentum. Moreover, selecting an accurate drug target is a critical task in finding agents with a particular biological activity that is anticipated to have therapeutic efficacy. Thus, role played by the NS2B/NS3 protein in the dengue virus life cycle is envisaged to form a potential attractive target for dengue inhibitors drug design. Coumarin analogues have received a considerable attention as a lead structure for the discovery of antiviral agents. Therefore, coumarin based thiadiazinyl hydrazones were designed as potential NS2B/NS3 protease inhibitors. Molecular docking studies provided insights into the binding modes of the inhibitors. All compounds were observed to have good interactions with the active site residues.
Yoko Matsumoto
Sojo University, Japan
Title: Therapeutic effects of trehalose liposomes against carcinoma along with apoptosis in vitro and in vivo
Time : 14:45-15:10

Biography:
Yoko matsumoto is a professor in the department of life sciences at Sojo University, Japan. She received her PhD in pharmacy from Kyushu University, Japan. She was a visiting researcher in Colorado University. She has received outstanding female researcher award from the society of chemical engineering, Japan. She is one of the directors for Japan nanomedicine society and councilor for japanese association for molecular target therapy of cancer. Her current research interest focuses on trehalose liposomes for therapeutic applications. She has published more than 120 papers in reputed journals.
Abstract:
Trehalose stabilizes membranes and proteins in cells most likely by hydrogen bonding. Trehalose liposomes (DMTre) composed of DMPC and trehalose micelles have been produced. Hydrodynamic diameter (dhy) of DMTre composed of 30 mol% DMPC and 70 mol% TreC14 was 100 nm with single and narrow range of size distribution, which was preserved for a period remaining stable for more than one month. The thickness of the fixed aqueous layer (TFAL) of DMTreCn was evaluated from the zeta potential and increase in TFAL values of DMTreCn was obtained in a dose-dependent manner. The TFAL values for DMTreCn were larger than that of DMPC liposomes. The remarkable inhibitory effects of DMTre on the growth of human colon, gastric, hepatocellular and lymphoblastic carcinoma cells have been reported. In this study, the inhibitory effects of DMTre on the growth of lung carcinoma (A-549) cells were examined in vitro and in vivo. DMTre inhibited the growth of A-549 cells leading to apoptosis. The activation of caspase-3, 8, and 9 was obtained for A-549 cells treated with DMTre. The suppression of tumor weight of xenograft mice model of carcinoma treated with DMTre was obtained. To investigate induction of apoptosis against tumor, the tissue section of tumor in
xenograft mice model of carcinoma after the treatment with DMTre was dyed and observed using microscope. Many apoptotic brown color cells in the tissue section of tumor was observed, indicating that DMTre could induce apoptosis in tumor cells against xenograft mice model of carcinoma.
John W M Yuen
The Hong Kong Polytechnic University, Hong Kong
Title: Quantification of some medical preparations of the plant origin
Time : 15:10-15:35

Biography:
John W M Yuen is currently an Associate Professor from the School of Nursing of the Hong Kong Polytechnic University. He is a biomedical scientist who has
completed his PhD in 2007, with a focus on cancer and immunology in the field of Urology. His research team conducts different types of research by adopting a
wide range of methodologies from exploratory cross-sectional/cohort design and in vitro laboratory experiments to in vivo trails on animals and humans.
Abstract:
Immunotherapeutic effects of the ethanol extract of Ganoderma lucidum (GLe) were compared against the conventional immunobladder® Bacillus Calmette-Guérin (BCG) in terms of cytotoxicity, cell cycle analysis and cytokine genes expression, in vitro. In conjunction with the intravesical study using the orthotopic MB49/C57 mice model, the murine urothelial carcinoma MB49 cell line was used for experiments. In agreement with the previous findings, GLe was demonstrated to exhibit G2/M phase cell arrest. On the other hand, dose-dependent cytotoxicity was demonstrated by both GLe and BCG as measured by the lactate dehydrogenase (LDH) assay; however, GLe concentrations ranged from 40 to 100 μg/ml killed 24.7-88.1% of the MB49 cells, which was superior to the 250-1000 μg/ml of BCG that killed 7.6-19.6%. Such cytotoxic effects were also shown to be inter-correlated with the expression of several cytokine genes, which are known to be important for anticancer. Although both GLe and BCG were shown to be active in inducing the interleukin(IL)-6, IL-12b and interferon-gamma (IFN-γ), dose-dependent inductions were only demonstrated by the range of GLe concentrations being tested. Particularly, the induction of IFN-γ gene was denominated by GLe up to 4-folded, as compared with the 1.5-folded increase by BCG. Basic research on immunobladder® BCG is limited and given that IFN-γ is wellevidenced for its anticancer effects, results here in speculated GLe could be an immunotherapeutic agent superior to the BCG by exerting stronger cytotoxic effects via a pathway involving IFN-γ and other molecules. The in vivo effects of GLe are currently being examined in animals.

Biography:
Abdulmajeed S H AlSamarrai was awarded a BSc in chemistry by the Mosul University of Iraq and MSc in analytical chemistry , by Baghdad University of Iraq and was awarded a PhD in organic chemistry by university of Birmingham/ UK in 1984. He is an assistant professor, department of chemistry, Tikrit university , Iraq (1991-2001). Then since 2001, he is a faculty member in the department of chemistry at the University of Samarra, Iraq and he is working as a professor of organic chemistry. Dr. ALSamarrai research interests are on organic synthesis of heterocyclic compounds and has published about 30 papers in reputed journals
Abstract:
This work includes the synthesis of some new 2-((4R,4aR,5aS,6S)-1,3-dioxo3,3a,4,4a,5,5a6,6a-octahydro-4,6-ethenocyclopropa[f]isoindol-2(1H)-yl)-N-(substituted phenyl) acetamides (3-11) by reaction of- (4R,4aR,5aS,6S)-1,3-dioxo3,3a,4,4a,5,5a6,6a-octahydro-4,6-ethenocyclopropa[f]isoindole1,3-(2H,3aH)-dione (1) with 2-chloro-N-(substituted phenyl) acetamides (2) in the presences of potassium carbonate in refluxing acetonitrile as a solvent.These compounds have been identified by spectroscopic Methods including, IR, 1H-NMR,13-NMR and elemental analysis (CHN).

Biography:
Zhongli Gao, Ph. D. is currently a Sr. Principal Scientist at Sanofi. He received his Ph. D. in Organic Chemistry at City University of New York in 1993. Upon graduation, he carried out his postdoctoral research on the total synthesis of Spinosyn A supervised by Professor Paquette at Ohio State University. He joined Hoechst Pharmaceuticals, one of the predecessor companies of Sanofi in 1995. He has worked on a broad range of disease areas in CNS, respiratory, inflammation, oncology, and rare disease involving GPCRs, proteases, enzymes, kinases, ion channels and transporters. He has led many projects in advancing compounds into clinical and preclinical or go/no-go decisions. In particular, his team has successfully transitioned SAR152954, a H3 receptor antagonist for Cognitive Impairment, and AVE8923, an orally active β-tryptase inhibitor for Asthma, into clinical development. He held 29 patents/ patent applications and 20+ peer reviewed publications.
Abstract:
Scaffold hopping is a technique that generates compounds containing a topologically different scaffold from the parent compound, but with similar or improved activity and other properties. This can be done intellectually by medicinal chemists or by computational algorithm. The technique is based on topological pharmacophore models developed from SAR of the current lead. In scaffold hopping, medicinal chemist’s insights into pharmacophore and SAR are crucial in this iterative process. Scaffold hopping can be used to generate new chemical entity; overcome patent or other limitations for the current leads; generate differentiated series of chemical matters. Common approaches include heterocycle replacements, bonds formation and cleavage, and topology-based hopping. This presentation will describe medicinal chemistry of H3 receptor antagonist and tryptase inhibitor programs via using scaffold hopping strategy to generate multi-genesis of chemical leads. Through optimization of the leads, clinical candidates were identified. The profile of the candidates and in vivo effects in disease animal models will also be briefly discussed.

Recent Publications (minimum 5)
- Sun H, Tawa G, Wallqvist A (2012) Classification of scaffold-hopping approaches. Drug Discovery Today 17: 310-324
- Hu Y, Stumpfe D, Bajorath J (2016) Recent Advances in Scaffold Hopping. J. Med. Chem.
- Gao Z, Hurst WJ, Hall D, Hartung R, Reynolds W, Kang J, Nagorny R, Hendrix JA, George PG (2015) Design and synthesis of a novel series of histamine H3 receptor antagonists through a scaffold hopping strategy. Bioorganic & Medicinal Chemistry. 23: 429–438
- Gao Z, Hurst WJ, et al., (2013) Synthesis, characterization, and biological assessment of the four stereoisomers of the H3 receptor antagonist 5-fluoro-2-methyl-N-[2-methyl-4-(2-methyl[1,3′]bipyrrolidinyl-1′-yl) phenyl] benzamide. Bioorganic & Medicinal Chemistry Letters. 23:4044–4047.
- Gao Z, Hurst WJ, et al., (2013) Identification and profiling of 3,5-dimethyl-isoxazole-4-carboxylic acid [2-methyl-4-((2S,3’S)-2-methyl-[1,30]bipyrrolidinyl-10-yl)phenyl] amide as histamine H3 receptor antagonist for the treatment of depression. Bioorganic & Medicinal Chemistry Letters. 23:6269–6273.
- Gao Z, Hurst WJ, et al., (2013) Discovery of a potent, selective, and orally bioavailable histamine H3 receptor antagonist SAR110068 for the treatment of sleep–wake disorders. Bioorganic & Medicinal Chemistry Letters. 23:6141–6145.
Jana Sopkova-de Oliveira Santos
University of Caen Normandy, France
Title: β-Strand Mimicry: Exploring Oligothienylpyridine Foldamers
Biography:
Jana Sopkova-de Oliveira Santos has completed his PhD at the age of 26 years from University Paris XI (Orsay)and University of Charles (Prague). Since 2012, she is a Professor of General Chemistry and Biophysic at University of Caen Normandy. She has published more than 110 papers in reputed journals.
Abstract:
Protein–protein interactions (PPIs) are involved in many cellular processes; consequently, the discovery of small molecules as modulators of PPIs has become a challenge in medicinal chemistry. Structural mimetics of protein secondary structures could maintain or restore biological functions and should possess biological activity. Actually, the most challenging classes of PPIs are those mediated by β-sheet, which are implicated in a number of diseases. Only a few β-strand mimics have been published to date. This study presents an evaluation of oligothienylpyridyl scaffolds in view of their ability for β-strand mimicry.
We have observed that a coplanar arrangement in thienylpyridyl systems can be obtained in several different ways. The presence of a nitogen and sulfur atom in the junction vicinity introduces a coplanar arrangement as well as the presence on the nitrogen atom in the non-ortho-substituted systems. The introduction of an ortho substituent in a system with a nitrogen atom in the junction vicinity perturbs the two rings somewhat, but the system can achieve the coplanar arrangement, because the energy barrier is very low. The same behavior was observed in a non-ortho-substituted biaryl with only a sulfur atom in the junction vicinity. The X-ray structures showed that the compounds have a tendency to adopt a nearly coplanar conformation and the positions of methyl substituents coincide well with those of i, i + 2nd or i, i + 4th β-strand side chains. Therefore, the thienylpyridine scaffold opens the way to produce coplanar compounds mimicking β-strand side-chain distributions.
Fabiola Porta
University of Basel, Switzerland
Title: Ezyme triggered nanomaterial for cancer therapy

Biography:
Dr. Fabiola Porta has studied Medicinal Chemistry and Pharmaceutical Technology in Milan at Universita’ degli studi, where she obtained her Master degree in Pharmacy in 2008. She then moved to the Leiden Institute of Chemistry where she graduated, in 2012, in chemistry with a special focus in nanoparticles synthesis and characterization. After the completion of her PhD she started to work as PostDoc in several institutions in Switzerland as: FHNW and University of Basel. Recently, she joined the group of Biopharmacy where she is developing-self assembled nanoparticles for cancer therapy. Dr. Porta expertise is in the design and development of nanovehicles for cancer therapy with aparticular focus on targeted nanocarriers for breast cancer therapy.
Abstract:
Polymer vesicles, are attracting much attention as alternative nanodelivery system to implement drug targeting strategies. Polymersomes have several interesting features. For instance, ease of chemical modification of the polymer chains can be used to modulate their tissue specificity and organ distribution. A wide variety of polymers is available, however a good candidate for pharmaceutical formulations is the di-block copolymer poly(dimethylsiloxane)-b-poly(2-methyloxazoline) (PDMS-PMOXA). This polymer is formed by two subunits which are FDA approved for the development of novel nanomaterials with a potential human use. In our work we are developing a responsive nanomaterial for the treatment of breast cancer. In order to develop a targeted delivery system a very good understanding of the cancer biology is necessary. In particular cancer biomarker are of particular interest due to their specificity for cancer cells. The enzyme family of cathepsins are highly expressed in certain type of cancers as breast tumor. Moreover, they are responsible for the degradation of proteins in the lysosomes. The elevate expression of these enzymes in tumors is an evidence of the increased metabolism of cancer cells. For this reason, these enzymes are a very interesting target for the development of a novel nanomaterial. In this work we are presenting a peptide cross linked polymeric nanoparticle with the main goal to encapsulate anticancer compounds and to release them only upon activation of the system by cathpesin B.
Recent Publications
- F. Porta and Alexander Kros, Colloidosomes as single implantable beads for the in vivo delivery of hydrophobic drugs, Particles & Particles Systems Characterization, 2013, 30, 606-613
- P. Nadrah, F. Porta, O. Planinšek, A. Kros, M. GaberšÄek , Poly(propylene imine) dendrimer caps on mesoporous silica nanoparticles for redox-responsive release: smaller is better, PCCP, 2013, 15, 10740-10748
- D. Witzigmann, S. Sieber, F. Porta, P. Grossen, A. Bieri, N. Strelnikova, T. Pfohl, C. Prescianotto-Baschong and J. Huwyler, Formation of lipid and polymer based gold nanohybrids using a nanoreactor approach, RCS Adv., 2015, 5, 74320
- D. Gliesche, J. Hussner, D. Witzigmann, F. Porta, T. Glatter, A. Schmidt, J. Huwyler, H. Meyer zu Schwabedissen, Secreted Matrix Metalloproteinase-9 of proliferating smooth muscle cells as a trigger for drug release from stent surface polymers in coronary arteries, Mol. Pharm., 2016, 13 (7), 2290-2300,
- D. Witzigmann, P. Detampel, F. Porta, J. Huwyler, Isolation of multiantennary N-glycans from glycoproteins for hepatocyte specific targeting via the asialoglycoprotein receptor, RCS Adv., 2016, 6, 97636-97640,
Hamid Irannejad
Mazandaran University of Medical Sciences, Sari, IRAN
Title: Synthesis and neuroprotective activity of novel 5,6-Diaryl-1,2,4-triazine derivatives with ethyl acetate moiety against H2O2 and Aβ-induced neurotoxicity

Biography:
Hamid Irannejad has completed his Pharm.D at Kerman University of Medical Sciences and PhD at Tehran University of Medical Sciences, IRAN. Postdoctoral studies was accomplished at University of Siena, Italy, under the supervision of Prof. Maurizio Botta. Currently, he is serving as an assistant professor at Mazandaran University of Medical Sciences. He has published nearly 20 papers in reputed journals in the field of medicinal chemistry.
Abstract:
Alzheimer’s disease (AD) is a neuropathologic disorder characterized by intracellular neurofibrillary tangles and amyloid aggregates in the CNS. In recent years numerous approaches have been used to combat AD like small molecule inhibitors of Aβ aggregation, anti-inflammatory agents, cholinesterase, β- and γ-secretase.
Herein, we report synthesis of some 5,6-diaryl-1,2,4-triazines 3a-f and 8a-e as potential agents for treatment of AD. We evaluated them against both H2O2 and β-amyloid induced toxicity in PC-12 and SH-SY5Y cells and the extent of cell viability and apoptosis were assessed.
The synthesis of compounds (3a-f) was started by 1,2-diketones, in which triazine ring closure was performed by thiosemicarbazide and alkylation by ethyl chloroacetate to afford compounds 3a-f.
synthetic route for compounds 8a-e was started by an acylation reaction of anisole with phenylacetic acid derivatives. The oximation in the alpha position of carbonyl group was performed by use of sodium methoxide and butylnitrite. The next two steps, were performed similarly to afford final compounds 8a-e.
All compounds showed significant neuroprotective activity with EC50 values ranging from 14-30 µM. Most compounds could increase cell viability compared to amyloid treated group.
Surprisingly, 3-thioxo-1,2,4-triazin-2(3H)-yl)acetate derivative 8e was the most potent compound in both tests with EC50 of 14 µM and could increase 40% of cell viability revealed by cytometric analysis with Annexin V/PI staining. It was also shown that 8e has more neuroprotective activity than Quercetin. Morphologic evaluation of cells by DAPI staining and TUNEL assay showed the effectiveness of this compound to improve neurite outgrowth in neuronal cells.
Yun Lu
Southern Illinois University Edwardsville, USA
Title: Mapping the Vibrational Transition-State Conformational Change in Enzymes for Drug Design

Biography:
Yun Lu received his Ph.D. degree in Organic Chemistry in 1996 from Nankai University, China. He did his postdoctoral work in Utah State University with Professor Vernon D. Parker and Wayne State University with Professor Martin Newcomb. His research focuses on the field of physical organic chemistry studying the mechanism of organic reactions and the transition state structures. He is now a professor in the Department of Chemistry at Southern Illinois University Edwardsville, USA. He has published nearly 50 papers in reputed journals.
Abstract:
About a quarter of the current registered pharmaceutical drugs are enzyme inhibitors. Many of them are the enzyme transition-state (TS) analogues that are designed largely on the basis of the crystal structure of the stable “frozen” TS analogues or the “still” TS structures from the kinetic studies. Recent decade has, however, witnessed much of the role of the enzyme dynamics in catalysis. Protein vibrations with different time scales have been proposed to assist various processes of the complex enzymatic reactions, including the bond-formation and - cleavage in the active site. The latter suggests a vibrational TS structure on the reaction coordinate, coupled with the local fast motions of enzyme. While study of the vibrational TS that involves the fast fluctuations of the reaction distance is still in its infancy, it would be worthy to consider the concept in design of what may come to be more effective inhibitors and more successful drugs. E.g., a successful inhibitor may be that can also interrupt the TS/enzyme coupled vibrations. In this paper, we present a method to gain the different TS structures at different donor-acceptor distance (DAD) in both solution and enzymatic H-transfer reactions. We use secondary (2°) kinetic isotope effect (KIE) as a structural descriptor. We determine the 2° KIEs and thus TS structures for hydride- and deuteride-tunneling processes that have different DADs. Information about the DAD-dependent TS structures in enzymes would help track the path of the vibrational TS conformational changes. This information would be useful for drug design.
S.I. Voychuk
Zabolotny Institute of Microbiology and Virology of NASU, Ukraine
Title: Morphological and structural features of adenovirus particles by reacting with bacterial cell

Biography:
Serhiy Voychuk has completed his PhD at the age of 26 years at Zabolotny Institute of Microbiology and Virology of NASU and continued his work in this institute. He is a senior researcher and a supervisor of a team of researchers with a broad spectrum of scientific interests in microbiology, virology, biotechnology and medicine. He is a coauthor of more than 40 papers
Abstract:
Antiviral drugs mainly targeted onto prevention of viral synthesis within the host cells or elimination of it enters into the cell through induction of natural defense mechanisms, among which are immune response and interferon synthesis. Recently, it was suggested that human microbiota may play a role in virus infection. Human viruses did not infect bacteria cells, but this does not exclude a possibility of bacteria participation in the virus propagation into the host cells.
An interaction of lactic acid bacteria with human adenovirus serotype5 (HAdV-C5) was studied. We used strains of bacteria some of which are normally can be found in human intestine, while others are supplied with various milk products: Lactobacillus plantarum (56 strains), Enterococcus spp. (23 strains), Leuconostoc spp. (14 strains), Lactococcuse lactis (3 strains) and Pediococcus spp. (1 strain). After 1h of interaction of viruses (VPs) with bacteria the samples were examined with electron microscopy.
In 17% of cases there were no VPs found that can suggest their total destruction, but in other cases the VPs were clearly seen performing well preserved viruses and viral proteins. The direct viral adhesion to the surface of bacteria was noticed for 23% of the strains, while in other cases the VPs were associated with extracellular matrix structures or as the free particles.
The current research is one of the first steps in understanding the role of microbiota in the virus infections development. It showed that various strains of the same bacterial species can cause opposite effects and lead both to the virus degradation or preservation and, therefore, can prevent or help virus to enter the human cells.
Daniel Ehrsam
University of Basel, Switzerland
Title: Early findings in the development of an enzymatically triggered nanoformulation

Biography:
Daniel Ehrsam studied Pharmaceutical Sciences and graduated at University of Basel in March 2014. He conducted his master thesis in research of brain ischemia pathways at Texas Tech Health Science Center under supervision of Dr. Margaret Weiss. He then worked at the Swiss Tropical and Public Health Institute on Helminth drug development (Prof. Dr. Jennifer Keiser). In August 2015 he joined the research group of Prof. Dr. Henriette E. Meyer zu Schwabedissen for his PhD studies. His research is focused on the development of an enzyme based targeted drug delivery system.
Abstract:
In order to spare healthy cells and decrease adverse effects, innovative concepts of tumor targeting aim at bringing the cytotoxic payload most selectively to tumor cells. One concept is to use enzymes overexpressed in the surrounding of proliferating cells, like the gelatinase matrix-metalloproteinase 9 (MMP-9), as a trigger for drug release.
Our aim is to synthesize and characterize self-assembling nanoformulations consisting of an MMP9-labile peptide coupled to an anti-cancer drug.
By use of bioconjugate chemistry an amphiphilic molecule containing paclitaxel and an MMP9-labile peptide was synthesized to form nanoparticles. To identify a tumor entity as a target for our novel nanoformulation we quantified expression of MMP-9 in a commercially available tissue collection by multiplex real-time PCR. Several tumor entities showed significantly increased expression comparing normal to malignant tissue. Immunohistochemistry and database analysis suggested brain tumors, particularly glioblastoma multiforme, as a tumor entity where MMP-9 could be used to trigger drug release. Established brain cancer cell lines were characterized for MMP-9 expression and activity. LN-18 and U87-MG cells were selected for in vitro characterization of the synthesized nanoformulation. In preparation of in vivo xenograft studies LN-18 and U87-MG cells were stably transfected with mKate2 and characterized for expression.
Taken together, we verified overexpression of MMP9 in glioblastoma multiforme. Commonly used brain cancer cell lines were characterized prior to in vitro studies on MMP9 triggered drug release, and preparations for in vivo xenograft studies have been finished. Further studies are warranted to fully characterize the nanoformulation and understand its effects in vivo.
- Medicinal Chemistry|Chemical Biology|Computer Aided Drug Design

Chair
Gaia Cecchi
University of Florence, Italy

Co-Chair
Monika I Konaklieva
American University, USA
Session Introduction
Concepción González-Bello
Universidade de Santiago de Compostela, Spain
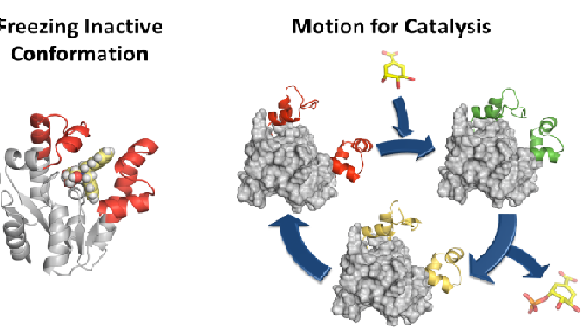
Title: Disabling essential enzyme motion for catalysis: An efficient approach for shikimate kinase inhibition
Time : 15:50-16:15

Biography:
Concepción González-Bello has obtained her PhD from the University of Santiago de Compostela (USC, Spain) in 1994. She did two Pre-doctoral stays at the University of Gent (Belgium) with professor vandewalle and at the Scripps Research Institute (USA) with professor Nicolaou. After a Post-doctoral stay at the University of Cambridge (UK) with professor Abell, she joined USC as an assistant professor, was promoted to associate professor in 2003 and obtained the Spanish habilitation to Full professor in 2011. She joined the Center for Research in Biological Chemistry and Molecular Materials (CIQUS) as a group leader in 2011. She is the author of about 80 papers and several book chapters and an Academic Editor of Plos One. Her main research interest is to develop updated therapies targeting infectious diseases, in particular, drugs with new mechanisms of action to combat the growth of antibiotic-resistant bacteria.
Abstract:
Although antibiotics are one of the most successful drugs in clinic that have saved millions of lives, many of them are nowadays ineffective in treating infections caused by resistant bacteria. It is therefore urgent to search for new antibacterial agents and approaches to face this huge challenge. Considering that most current antibiotics that are highly successful in human clinical use, targeted at only four main key processes and resistance to these antibiotics is widespread and well known, the search for unexplored bacterial functions appears to be a good option for the development of novel antimicrobial agents with a new mechanism of action. Our group is studying the possible development of new antibiotics by the selective and effective inhibition of an essential enzyme in bacteria that does not have any counterpart in human cells, shikimate kinase (SK, aroK gene). In particular, we are focused on SK from M. tuberculosis and H. pylori, two important pathogenic bacteria. Based on the essential enzyme motion for catalysis and product release studied by molecular dynamics simulation studies, potent reversible competitive inhibitors of the enzyme were developed. Compounds that stabilize the closed conformation for catalysis or the open conformation for product release were developed. An ester prodrug approach was used for achieving good in vitro activities against H. pylori. Our results also show that the less exploited motion-based design approach, not only is an alternative strategy for the development of competitive inhibitors, but could also be a way to achieve selectivity against a particular enzyme among its homologous ones. By using this approach, (1) the presence in the selected pocket of residues with markedly different properties would not be required, and (2) the effects of changes on residues to avoid the inhibition (resistance) should have a less pronounced effect.
Recent Publications
1.Blanco B, Prado V, Lence E, Otero J M, Garcia-Doval C, van Raaij M J, Llamas-Saiz A L, Lamb H, Hawkins A R, González-Bello C (2013) Mycobacterium tuberculosis shikimate kinase inhibitors: Design and simulation studies of the catalytic turnover. J. Am. Chem. Soc. 135: 12366-12376.
2.Prado V, Lence E, Maneiro M, Vázquez-Ucha J C, Beceiro A, Thompson P, Hawkins A R, González-Bello C (2016) Targeting the motion of shikimate kinase: Development of competitive inhibitors that stabilize an inactive open conformation of the enzyme. J. Med. Chem. 59: 5471-5487.
3.Prado V, Lence E, Thompson P, Hawkins A R, González-Bello C (2016) Freezing the dynamic gap for selectivity - motion-based design of inhibitors of the shikimate kinase enzyme. Chem. Eur. J. 22: 17988-18000.
4.Prado V, Lence E, Vallejo J A, Beceiro A, Thompson P, Hawkins A R, González-Bello C (2016) Study of the Phosphoryl-transfer mechanism of shikimate kinase by NMR spectroscopy. Chem. Eur. J. 22: 2758-2768.
5.González-Bello C (2016) Inhibition of shikimate kinase and type II dehydroquinase for antibiotic discovery: Structure-based design and simulation studies. Curr. Top Med. Chem. 16: 960-977.
Fabiola Porta
University of Basel, Switzerland
Title: Enzyme triggered nanomaterial for cancer therapy
Time : 16:15-16-40

Biography:
Fabiola Porta has studied Medicinal Chemistry and Pharmaceutical Technology in Milan from the Universita’ degli studi, where she obtained her Master’s degree in Pharmacy in the year 2008. She then moved to the Leiden Institute of Chemistry, where she graduated in Chemistry with a special focus on nanoparticles synthesis and characterization in the year 2012. She has done her post-doctoral studies from FHNW and University of Basel. Recently, she joined the group of Bio-pharmacy, where she is developing-self assembled nanoparticles for cancer therapy. She has expertise in the design and development of nano-vehicles for cancer therapy with a particular focus on targeted nanocarriers for breast cancer therapy.
Abstract:
Polymer vesicles are attracting much attention as alternative nano-delivery system to implement drug targeting strategies. Polymersomes have several interesting features. For instance, ease of chemical modification of the polymer chains can be used to modulate their tissue specificity and organ distribution. A wide variety of polymers is available, however a good candidate for pharmaceutical formulations is the di-block copolymer poly(dimethylsiloxane)-b-poly(2methyloxazoline) (PDMS-PMOXA). This polymer is formed by two subunits which are FDA approved for the development of novel nanomaterials with a potential human use. In our work, we are developing a responsive nanomaterial for the treatment of breast cancer. In order to develop a targeted
delivery system a very good understanding of the cancer biology is necessary. In particular cancer biomarker are of particular interest due to their specificity for cancer cells. The enzyme family of cathepsins is highly expressed in certain type of cancers as breast tumor. Moreover, they are responsible for the degradation of proteins in the lysosomes. The elevate expression of these enzymes in tumors is an evidence of the increased metabolism of cancer cells. For this reason, these enzymes are a very interesting target for the development of a novel nanomaterial. In this work, we are presenting a peptide cross linked polymeric nanoparticle with the main goal to encapsulate anticancer compounds and to release them only upon activation of the system by cathepsin B.
Recent Publications
1.Blanco B, Prado V, Lence E, Otero J M, Garcia-Doval C, van Raaij M J, Llamas-Saiz A L, Lamb H, Hawkins A R, González-Bello C (2013) Mycobacterium tuberculosis shikimate kinase inhibitors: Design and simulation studies of the catalytic turnover. J. Am. Chem. Soc. 135: 12366-12376.
2.Prado V, Lence E, Maneiro M, Vázquez-Ucha J C, Beceiro A, Thompson P, Hawkins A R, González-Bello C (2016) Targeting the motion of shikimate kinase: Development of competitive inhibitors that stabilize an inactive open conformation of the enzyme. J. Med. Chem. 59: 5471-5487.
3.Prado V, Lence E, Thompson P, Hawkins A R, González-Bello C (2016) Freezing the dynamic gap for selectivity - motion-based design of inhibitors of the shikimate kinase enzyme. Chem. Eur. J. 22: 17988-18000.
4.Prado V, Lence E, Vallejo J A, Beceiro A, Thompson P, Hawkins A R, González-Bello C (2016) Study of the Phosphoryl-transfer mechanism of shikimate kinase by NMR spectroscopy. Chem. Eur. J. 22: 2758-2768.
5.González-Bello C (2016) Inhibition of shikimate kinase and type II dehydroquinase for antibiotic discovery: Structure-based design and simulation studies. Curr. Top Med. Chem. 16: 960-977.
Monika I Konaklieva
American University, USA
Title: A new class of b-lactam antibiotics active against drug-sensitive and drug-resistant Mycobacterium tuberculosis
Time : 16:40-17:05

Biography:
Monika I Konaklieva has completed her PhD in Chemistry from SUNY Buffalo in 1997, and became a visiting professor in Medicinal Chemistry at Midwestern University, Chicago, Illinois (1997-1999). She is currently an associate professor at the American University. She has published more than 40 papers in reputed journals and has been serving as an Editorial Board Member of several chemistry journals publishing in the areas of organic and medicinal chemistry.
Abstract:
We have designed, synthesized, and tested monocyclic β-lactams that carry aryl-thioether group at C4. These lactams have shown good intrinsic activity against serine β-lactamase producing Mycobacterium tuberculosis H37Rv (Mtb). Some of the compounds have demonstrated minimal inhibitory concentration (MIC) as low as 6.25 μg/ml in 7H9 and 0.19 μg/ml in GAST. Our investigations indicate that these compounds are cidal to both replicating and non-replicating persistent Mtb. These compounds have also shown activity against multi-drug resistant strains of M. tuberculosis. Therefore, they are promising candidates for lead discovery. Mechanism of action and target identification studies are currently underway.
Michele Tonelli
University of Genoa, Italy
Title: Host DHFR-directed cycloguanil analogues endowed with promising activity against influenza virus and respiratory syncytial virus
Time : 17:05-17:30

Biography:
Michele Tonelli has done his graduation in chemistry and pharmaceutical technology from Genoa University in the year 2003. He did his PhD in pharmaceutical food and cosmetic sciences. He has participated in many national research projects. Since 2012, he is an Assistant Professor (Medicinal Chemistry) in the Department of Pharmacy, University of Genoa. He has published more than 25 papers in reputed journals and has been serving as Reviewer of Journal of Medicinal Chemistry, Bioorganic & Medicinal Chemistry, Antiviral Research, Future Medicinal Chemistry, etc.
Abstract:
The Orthomyxoviridae and Paramyxoviridae families comprise important respiratory pathogens, such as respiratory syncytial virus, influenza A and B viruses. The acute respiratory illnesses caused by these viruses represent a major medical need. In particular, the risk for a severe influenza A pandemic leads to the development of new and broadly acting therapeutics an urgent issue. Currently, used antiviral drugs preferentially inhibit virus-specific replication factors. An alternative and emerging strategy is to address host factors involved in virus replication, because less susceptible to mutate. Herein, we have identified a series of 4,6-diamino- 1,2-dihydrotriazines, structurally related to the antimalarial drug cycloguanil, as new inhibitors of influenza A and B virus and RSV via targeting of the host dihydrofolate reductase (DHFR) enzyme. They proved active against influenza B virus in the low micromolar
range, sometimes reaching the sub-micromolar potency of zanamivir (EC50=0.060 μM), and markedly exceeded (up to 327 times) the antiviral efficacy of ribavirin. Besides inhibiting two influenza A strains, more importantly the compounds displayed nanomolar activity against RSV with a SI (CC50/EC50)>10,000 for the most active compounds (EC50~0.008 μM), far surpassing the potency and safety profile of the licensed drug ribavirin (EC50=5.8 μM, SI>43). These compounds, tested against the recombinant protein of the hDHFR, also confirmed to bind this enzyme in the sub-micromolar range. Kinetic inhibition studies showed a competitive inhibition behavior, and docking studies disclosed the most probable binding mode for this class of compounds as hDHFR ligands.